Dive into the intricate realm of single-cell RNA sequencing (scRNA-Seq) for profound insights into cellular processes. This blog delves into the basics, methodology, and applications of scRNA-Seq, enriched by recent research findings.
Refining Data: Preprocessing and Noise Control
Start with tools like Cutadapt and Trimmomatic to eliminate adapter sequences and enhance data quality. SinQC identifies gene expression outliers by assessing sequencing library quality, effectively handling data noise.
Quantifying Features and Filtering Genes
Translate alignment results into gene expression profiles through feature quantification. Widely used tools like RSEM and Cufflinks predict RNA-Seq sample reads using statistical models. Alternatively, UMIs directly track transcripts, offering distinct advantages. To manage scRNA-Seq’s substantial noise, gene filtering (FPKM, UMI counts) retains pertinent genes for analysis.
Confronting Challenges: Addressing Confounders and Analysing Expression Differences
Mitigate batch effects that cause systematic shifts using methods like COMBAT and BASICS. This enhances downstream analysis robustness. For vital gene expression changes, differential expression analysis employs approaches like MAST, ZIFA, and BISCUIT. Graph-based methods, including kNNG comparison and SNN graphs, unveil cell lineages and clusters within scRNA-Seq datasets.
Unveiling Microevolution and Future Horizons
Tools like Monocle and Embeddr enable microevolution studies without spatial/temporal cues. They reveal cell temporal order and gene expression dynamics. Incorporating spatial/temporal data in microevolution models provides fresh insights into gene expression dynamics and stem cell regulatory networks. Applications extend to tumour heterogeneity and cancer evolution, comprehending intricate processes.
scRNA-Seq: Catalyst for Advancement
In the evolving landscape of single-cell genomics, scRNA-Seq emerges as a revolutionary tool. Overcoming challenges, from refining protocols to expanding analyses, remains crucial. As scRNA-Seq data volume grows, innovative tools emerge, offering deeper insights and novel avenues. Researchers exploit scRNA-Seq’s potential to decode biological complexity by blending foundational knowledge with cutting-edge methods.
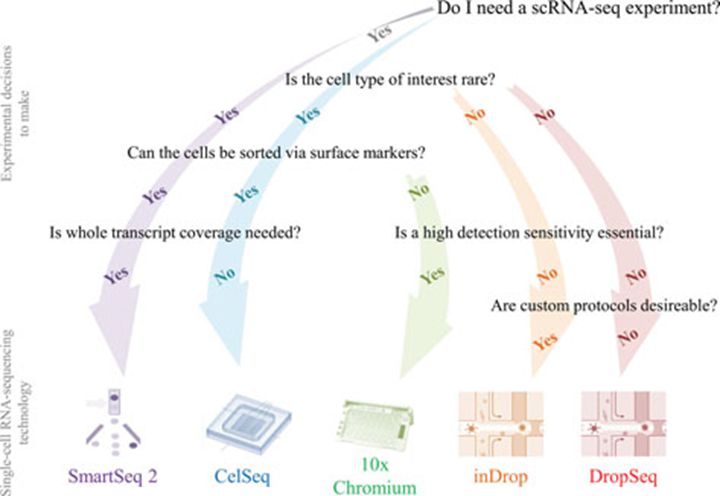
Figure 1. Flow chart guide to the most suitable scRNA-seq technique depending on the scientific problem and the boundary conditions.
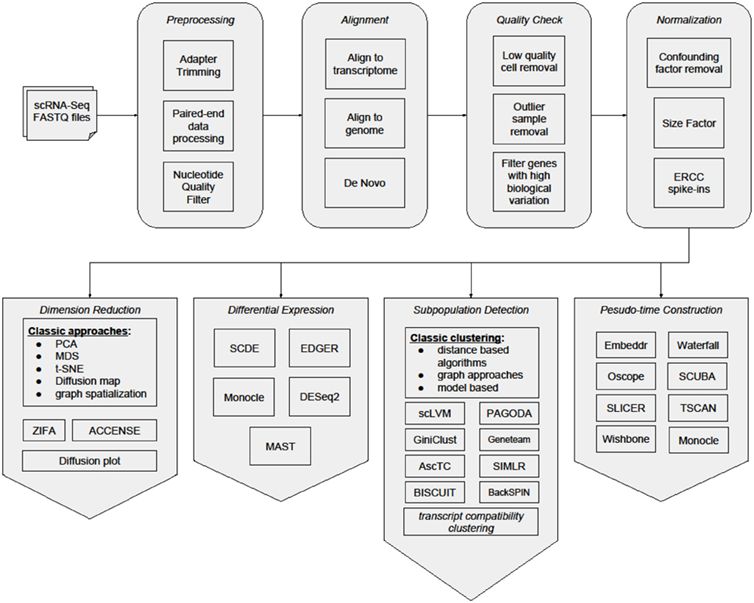
Figure 2. Workflow of single cell RNA-seq analysis
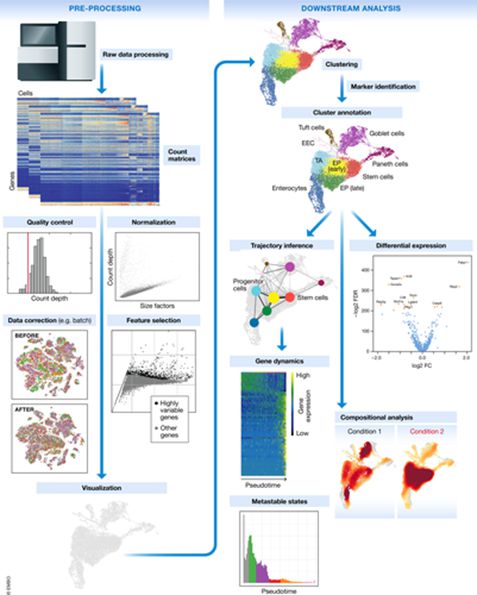
Figure 3. Schematic of a typical single-cell RNA-seq analysis workflow
Malte D Luecken, Fabian J Theis. Mol Syst Biol. (2019) 15: e8746
CODE
| library(Seurat) # Load scRNA-Seq data (Assuming you have a count matrix) # Replace ‘counts_matrix_dromics.csv’ with your actual data file counts <- read.csv(‘counts_matrix_dromics.csv’, row.names = 1) # Create a Seurat object sc_data <- CreateSeuratObject(counts = counts) # Quality control (filter out low-quality cells and genes) sc_data <- FilterCells(sc_data, subset.names = c(‘nGene’, ‘nUMI’), low.thresholds = c(200, 500), high.thresholds = c(6000, 30000)) sc_data <- FilterGenes(sc_data, min.cells = 3) # Normalize data sc_data <- NormalizeData(sc_data) # Identify variable genes sc_data <- FindVariableFeatures(sc_data, selection.method = ‘vst’, nfeatures = 2000) # Scaling and centering sc_data <- ScaleData(sc_data) # Perform PCA sc_data <- RunPCA(sc_data, features = VariableFeatures(object = sc_data)) # Perform UMAP visualization sc_data <- RunUMAP(sc_data, reduction = ‘pca’, dims = 1:10) # Cluster cells sc_data <- FindNeighbors(sc_data, reduction = ‘pca’, dims = 1:10) sc_data <- FindClusters(sc_data, resolution = 0.5) # Visualize results DimPlot(sc_data, label = TRUE) |
MCQs:
- What is the primary purpose of single-cell RNA sequencing (scRNA-Seq)?
- Identifying adapter sequences
- Enhancing data quality
- Exploring cellular processes at a single-cell level
- Analysing bulk RNA samples
Ans. 3. Exploring cellular processes at a single-cell level
- How do methods like COMBAT and BASICS contribute to scRNA-Seq analysis?
- Predict RNA-Seq sample reads
- Detect gene expression outliers
- Reduce batch effects for more robust analysis
- Filter genes based on FPKM values
Ans. 3. Reduce batch effects for more robust analysis
- In the field of single-cell genomics, what role does scRNA-Seq play?
- Analysing bulk RNA samples
- Decoding biological complexity at the single-cell level
- Enhancing the quality of sequencing libraries
- Removing noise from gene expression data
Ans. 2. Decoding biological complexity at the single-cell level


