
Medicine improves the quality of life by preventing or treating diseases. The fate of a drug from its discovery to launch into the market involves multifaceted complicated junctures. The drug development (DD) process involves four crucial phases: discovery, preclinical studies, clinical development and market approval (Hughes et al., 2011; Velmurugan et al., 2020). The search for a novel drug target, identification of compounds with drug like characteristics that passes clinical development leading to clinical trials demands enormous time and labour intensive tasks. Computer Aided Drug Design (CADD) offers indispensable strategies to address the shortcomings in each phase of the drug discovery and development pipeline. CADD offers assistance in the time taking initial phase of drug discovery and development thereby decreasing the duration and cost required to perform initial screening (Zhang et al., 2022) .
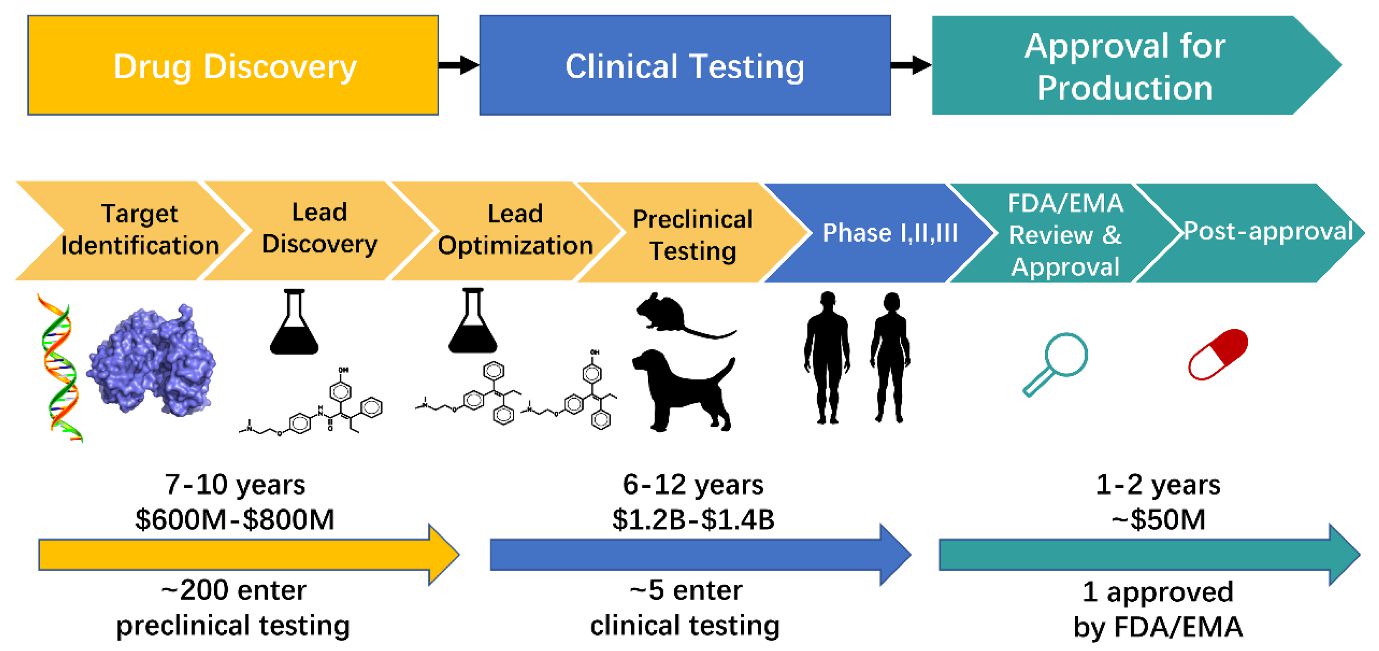
Figure 1. The process of drug research and development (Zhang et al., 2022).
The target-driven discovery phase starts by choosing a single or multiple drug target. The drug target must be disease-modifying and druggable. The active scaffolds (ligands) which shows some activity towards the target are chemically modified to mine out potential candidates. Virtual screening (VS) techniques whether used as standalone or together with High throughput screening is an important tool in DD. VS utilizes computational approaches to screen drug-like compounds from diverse and vast collection of compound libraries. VS uses either the knowledge about the target protein structure (structure based VS) or the knowledge about known bioactive compounds (ligand based VS). Molecular docking is prominently used technique of structure-based VS (Kontoyianni, 2017). Quantitative Structure Activity Relationship (QSAR) follows ligand based VS approach.
The systematic screening of the compound libraries leads to the identification of hits. The actual drug design process starts here, where chemical modifications are introduced in the active scaffolds to optimize the interaction with the target in order to gain potency, and/or selectivity towards the target. The application of in silico absorption, distribution, metabolism, excretion, and toxicity (ADMET) filters in the early stages of DD will eliminate low affinity ligands (Bergström et al., 2016). The filters greatly rely on distribution analysis of physicochemical properties of drugs. The inclusion of these filters have reduced ADMET related project failures.
The contribution of CADD in the drug discovery is enormous with the continuous advancement in computational power, deep learning, machine learning and big data. Though CADD has reduced many attritions care should be taken while implementing these techniques, care should be taken while implementing automation. The final assessment by different professionals involved in the drug discovery cycle at each stage of the drug is crucial before entering into the clinical trial phase.
References
- Bergström, C. A. S., Charman, W. N., & Porter, C. J. H. (2016). Computational prediction of formulation strategies for beyond-rule-of-5 compounds. Advanced Drug Delivery Reviews, 101, 6–21.
- Hughes, J. P., Rees, S. S., Kalindjian, S. B., & Philpott, K. L. (2011). Principles of early drug discovery. British Journal of Pharmacology, 162(6), 1239.
- Kontoyianni, M. (2017). Docking and Virtual Screening in Drug Discovery. Methods in Molecular Biology (Clifton, N.J.), 1647, 255–266.
- Velmurugan, D., Pachaiappan, R., & Ramakrishnan, C. (2020). Recent Trends in Drug Design and Discovery. Current Topics in Medicinal Chemistry, 20(19), 1761–1770.
- Zhang, Y., Luo, M., Wu, P., Wu, S., Lee, T. Y., & Bai, C. (2022). Application of Computational Biology and Artificial Intelligence in Drug Design. International Journal of Molecular Sciences, 23(21).


