Drug discovery and development is a complex and time-consuming process. Bringing a new drug to the market typically takes over a decade. Researchers face challenges in screening potential drug molecules from a vast chemical space. Traditional methods, such as computational simulations and chemical-biological experiments, are resource-intensive and time-consuming. In recent years, computational approaches have gained prominence in predicting drug-target interactions (DTIs) more efficiently and rapidly.
The Importance of DTI Prediction
Predicting DTIs is a crucial preliminary step in drug development. It helps researchers identify potential drug candidates and assess their safety, efficacy, and rationality. By using computational methods, scientists can focus on target molecules selected through computer-aided screening. These predictions accelerate drug development cycles and reduce reliance on expensive and lengthy in vitro and in vivo experiments.
Categories of Computational Methods
Three major categories of computational methods exist for predicting DTIs:
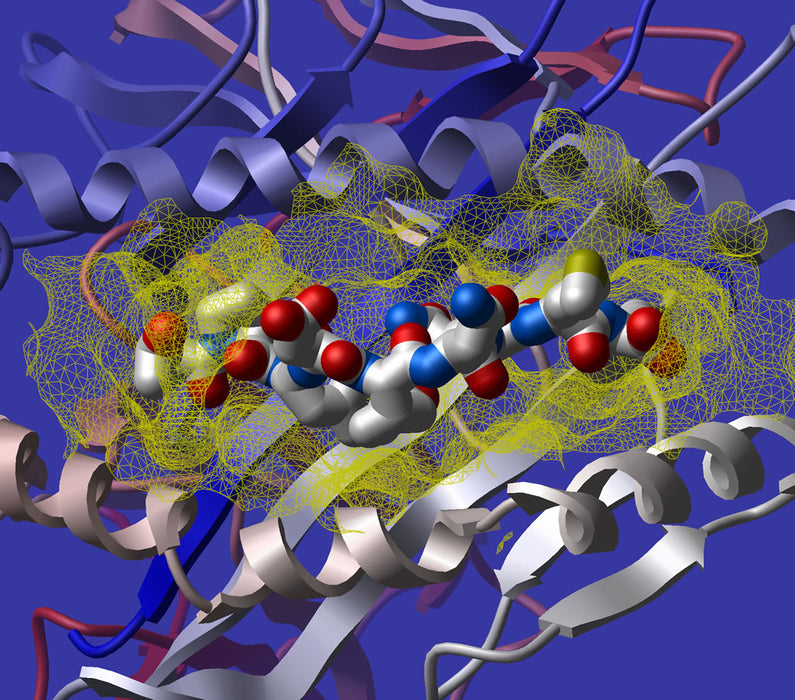
- Docking Simulation: Docking simulations predict how drug molecules interact with target proteins at a molecular level. These simulations aim to find the preferred bound complex of a given system. By understanding the binding affinity between a drug and its target, researchers can assess potential interactions and optimize drug design.
- Ligand-Based Methods: These methods rely on known ligand structures to predict interactions. They analyze similarities between ligands and identify potential targets based on shared features. Ligand-based approaches are useful when high-quality data for drug molecules and proteins are available.
- Chemogenomics Methods: These methods integrate genomic and chemical information to predict interactions. By considering genetic and chemical features, researchers can identify potential drug targets and optimize drug discovery.
Deep Learning and Data Representation
Deep learning techniques have revolutionized DTI prediction. Researchers use neural network models to learn complex patterns from data. Key aspects include:
- Data Representation: Deep learning-based methods use various representations for drugs and proteins. These include molecular fingerprints, graph-based representations, and embeddings. Proper data representation is critical for accurate predictions.
- Taxonomy of Deep Neural Network Models: Researchers have developed several deep neural network architectures for DTI prediction. These models include graph convolutional networks (GCNs), recurrent neural networks (RNNs), and attention-based models. Each architecture has its strengths and limitations.
Datasets and Evaluation Metrics
Commonly used datasets and evaluation metrics facilitate hands-on practice in DTI prediction. Researchers rely on curated datasets containing known drug-target pairs. Evaluation metrics, such as precision, recall, and area under the receiver operating characteristic curve (AUC-ROC), help assess model performance.
In conclusion, docking simulations play a pivotal role in predicting drug-target interactions. As AI technology continues to advance, machine learning remains an indispensable tool for efficient and accurate drug development. By leveraging computational methods, researchers can unlock new therapeutic possibilities and accelerate the discovery of life-saving medications.
References:
- Shi, W., Peng, D., Luo, J., Chen, G., Yang, H., Xie, L., Yin, X.-X., & Zhang, Y. (2023). A Review on Predicting Drug Target Interactions Based on Machine Learning. Lecture Notes in Computer Science, 14305, 318–333.
- Molecular Docking: Shifting Paradigms in Drug Discovery. International Journal of Molecular Sciences, 20(18), 4331.


